ABOUT
Research work in the Molecular Thermodynamics and Modeling of Materials Laboratory (MTMML) focuses on the development and implementation of novel hierarchical methods and algorithms for the computer modelling and calculation of advanced material properties at the molecular, mesoscopic and macroscopic levels (see Figure 1). Through this work, quantitative links are established between chemical constitution, processing conditions, and properties (physical, mechanical, rheological, transport, interfacial, optical, dielectric etc), which are critical for the optimal design of industrial processes and also govern the end-use performance of commercial products. In parallel, the molecular mechanisms underlying structure – property – processing – performance relations are elucidated with the objective of designing new, tailor-made materials.
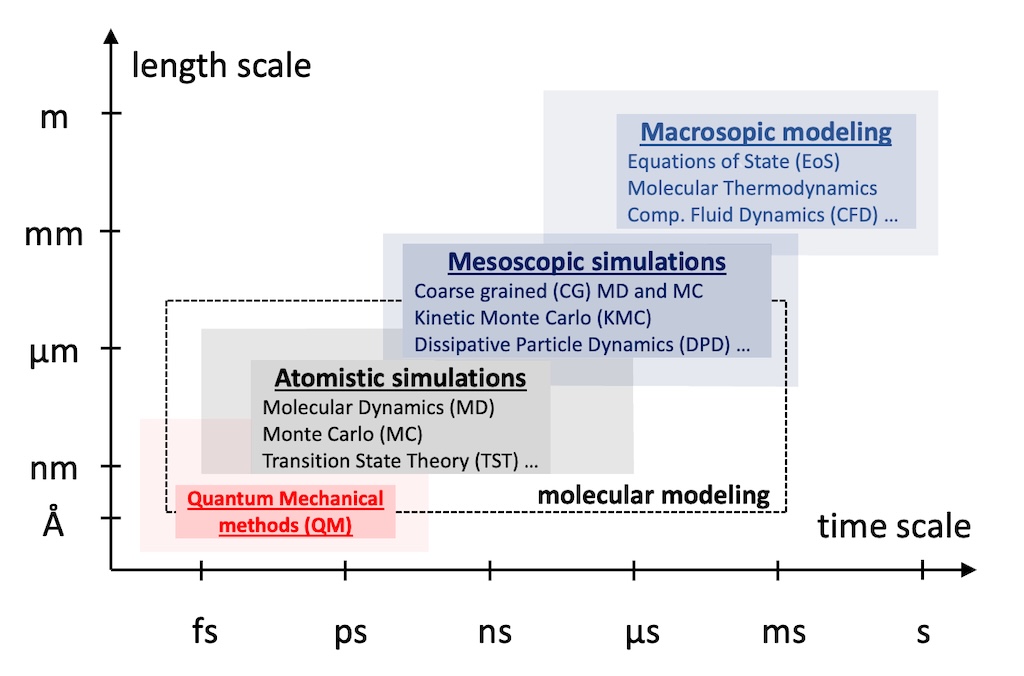
The hierarchical approaches developed and implemented at MTMML start with atomistic simulations addressing length scales on the order of tens of nanometers and time scales on the order of tens of nanoseconds (e.g., Monte Carlo, molecular dynamics, transition-state theory analysis of infrequent events) and proceed with mesoscopic methods (e.g., entanglement network modeling, kinetic Monte Carlo simulation, self-consistent field theory of inhomogeneous systems) to address longer time- and length scale phenomena. Finally, for the efficient design of novel processes mainly for the chemical, polymer and pharmaceutical industry, accurate macroscopic models, mostly in the form of equations of state (eos), are developed for phase equilibria and other thermodynamic properties of multicomponent mixtures. These eos are rooted to statistical mechanics and can be safely extrapolated to conditions where limited or no experimental data exist.