Bioinformatics
The complexity of the primary structure of human DNA is explored using methods from nonequilibrium statistical mechanics, dynamical systems theory and information theory. A collection of statistical analyses are performed on the DNA data and the results are compared across species and with sequences derived from different stochastic processes. Although detailed balance seems to hold at the level of a binary alphabet in genomic data, it fails when all four basepairs are considered, suggesting spatial asymmetry and irreversibility. Furthermore, the block entropy does not increase linearly with the block size, reflecting the long range nature of the correlations in the human genomic sequences. To probe locally the spatial structure of the DNA sequence we study local quantities, such as the exit distances from a specific symbol, the distribution of recurrence distances and the Hurst exponent, all of which show power law tails and long range characteristics. These results suggest that human DNA can be viewed as a non-equilibrium structure maintained in its state through interactions with a constantly changing environment.
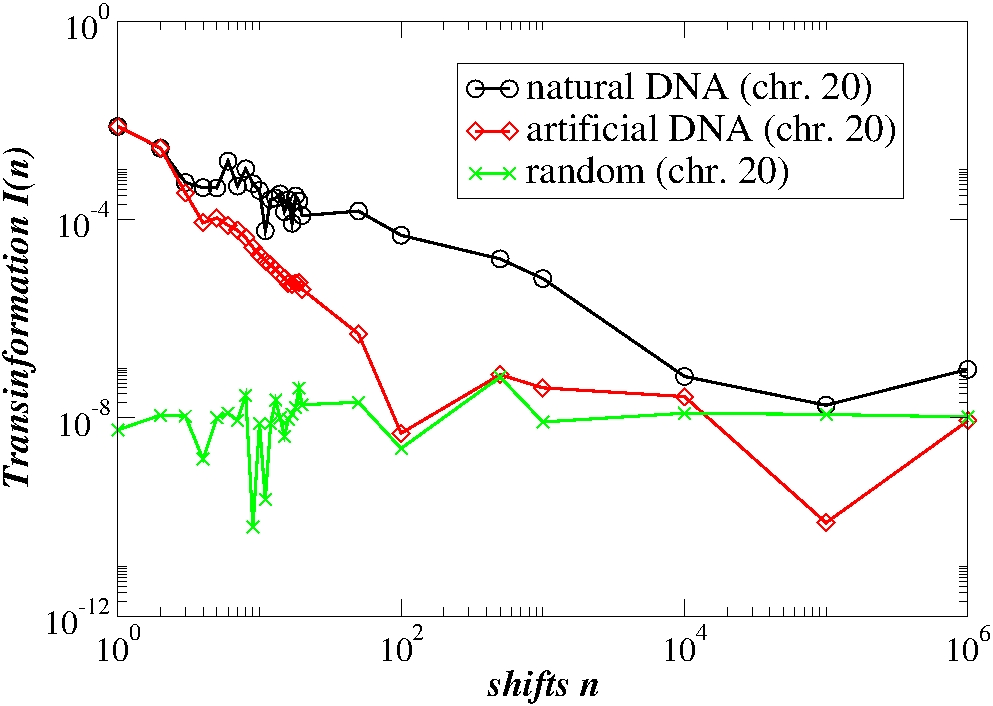
Complexity measures are also used to compare the genomic characteristics of different organisms belonging to distinct classes spanning the evolutionary tree: higher eukaryotes, amoebae, unicellular eukaryotes and bacteria. We have demonstrated that the conditional probability matrix for the four-letter and AT-CG alphabet is markedly asymmetric in eukaryotes while it is nearly symmetric in bacterial genomes. Overall, the conditional probability, the fluxes, the block entropy content and the exit distance distributions can be used as markers to discreminate functional groups in DNA sequences.